Analysis of 10x Visium mouse brain slice#
In this tutorial, we demonstrate SpaSRL on the analysis of 10x Visium Mouse Brain Serial Section 1 (Sagittal-Anterior) slice including
Self-representation learning
Spatial domain identification
Spatial domain annotation
Finding differentially expressed genes
The dataset is available at 10x genomics website (Spatial Gene Expression >> Visium Demonstration (v1 Chemistry) >> Space Ranger 1.0.0 >> Mouse Brain Serial Section 1 (Sagittal-Anterior)).
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
import matplotlib.pyplot as plt
import SpaSRL
%matplotlib inline
Data loading and preprocessing#
We load the dataset and perform preprocessing including finding highly variable genes and log transformation.
[2]:
adata = sc.read_visium('./data/Mouse Brain Serial Section 1 (Sagittal-Anterior)/')
adata.var_names_make_unique()
adata
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
Variable names are not unique. To make them unique, call `.var_names_make_unique`.
[2]:
AnnData object with n_obs × n_vars = 2696 × 31053
obs: 'in_tissue', 'array_row', 'array_col'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'spatial'
obsm: 'spatial'
[3]:
sc.pp.highly_variable_genes(adata, n_top_genes=2000, flavor='seurat_v3')
sc.pp.log1p(adata)
Self-representation learning#
We select landmark samples and then use selected landmarks to run self-representation learning.
[4]:
SpaSRL.select_landmarks(adata, n_landmarks=1000)
84%|████████████████████████████████████████████▎ | 1670/2000 [00:54<00:10, 30.42it/s, seleted landmarks: 1000]
[5]:
SpaSRL.run_SRL(adata, Lambda=0.1, n_neighbors=200)
21%|██████▍ | 107/500 [00:02<00:07, 52.68it/s, relChg: 2.714e-05, recErr: 9.931e-06, converged!]
This is how the adata looks like after self-representation learning.
[6]:
adata
[6]:
AnnData object with n_obs × n_vars = 2696 × 31053
obs: 'in_tissue', 'array_row', 'array_col', 'is_landmark'
var: 'gene_ids', 'feature_types', 'genome', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'spatial', 'hvg', 'log1p', 'select_landmarks', 'representation'
obsm: 'spatial'
obsp: 'representation'
Spatial domain identification#
We identify spatial domains using the representation matrix.
[7]:
sc.tl.leiden(adata, resolution=2.6, neighbors_key='representation')
[8]:
sc.pp.pca(adata)
sc.tl.umap(adata, init_pos='random', neighbors_key='representation')
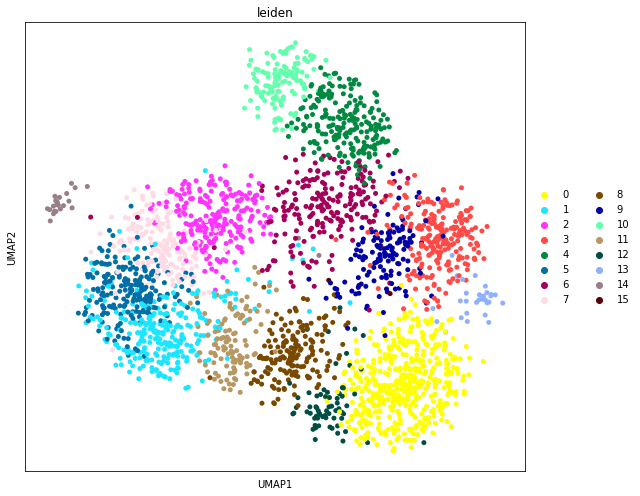
We visualize the inferred spatial domains in UMAP space.
[9]:
fig, axs = plt.subplots(figsize=(9, 7))
sc.pl.umap(
adata,
color="leiden",
size=100,
palette=sc.pl.palettes.default_102,
legend_loc='right margin',
show=False,
ax=axs,
)
plt.tight_layout()
C:\ProgramData\Anaconda3\lib\site-packages\anndata\_core\anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Reordering categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'feature_types' as categorical
C:\ProgramData\Anaconda3\lib\site-packages\anndata\_core\anndata.py:1220: FutureWarning: The `inplace` parameter in pandas.Categorical.reorder_categories is deprecated and will be removed in a future version. Reordering categories will always return a new Categorical object.
c.reorder_categories(natsorted(c.categories), inplace=True)
... storing 'genome' as categorical
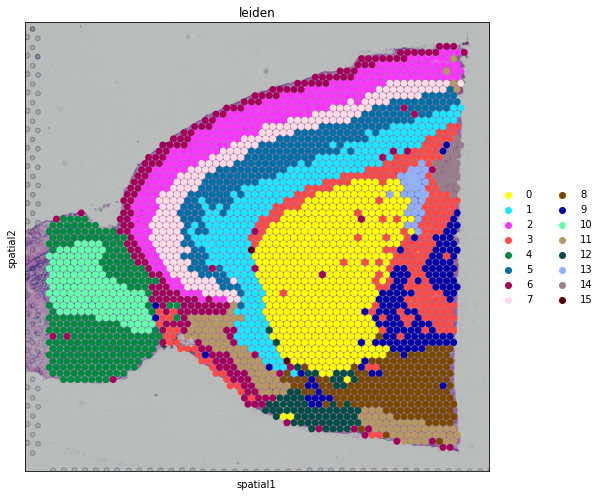
We also visualize the inferred spatial domains in spatial coordinates.
[10]:
fig, axs = plt.subplots(figsize=(9, 7))
sc.pl.spatial(
adata,
img_key="hires",
color="leiden",
size=1.5,
palette=sc.pl.palettes.default_102,
legend_loc='right margin',
show=False,
ax=axs,
)
plt.tight_layout()
Spatial domain annotation#
After spatial domain identification, we compute the differentially expressed (DE) genes of each domain for annotation.
[11]:
sc.tl.rank_genes_groups(adata, groupby='leiden', method='t-test')
[12]:
marker_genes = pd.DataFrame(adata.uns['rank_genes_groups']['names']).iloc[:10,:]
marker_genes
[12]:
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Ppp1r1b | 3110035E14Rik | Camk2n1 | Plp1 | Calb2 | 1110008P14Rik | Ptgds | Stx1a | Zcchc12 | Mobp | Gng4 | Nptxr | Tac1 | Ttr | Neurod6 | Scarf1 |
| 1 | Gpr88 | Slc17a7 | Camk2a | Mobp | Doc2g | Scn1b | Myoc | Nrn1 | Nap1l5 | Mbp | Gpsm1 | Olfm1 | Ppp1r1b | Enpp2 | Camk2a | 1700102H20Rik |
| 2 | Pde10a | Ttc9b | Lamp5 | Mbp | Fabp7 | Cck | Mgp | Camk2n1 | Bc1 | Plp1 | Pcbp3 | Ttc9b | Rgs9 | 1500015O10Rik | Cabp7 | Rin3 |
| 3 | Rgs9 | Nptx1 | Cck | Mal | Slc6a11 | Slc17a7 | Gjb2 | Slc17a7 | Gaa | Trf | Synpr | Slc17a7 | Gad2 | Cd9 | Nsmf | Zap70 |
| 4 | Penk | Ncald | Nptxr | Trf | Cdhr1 | Vsnl1 | Aebp1 | Cck | Nrsn2 | Agt | Meis2 | Ndrg3 | Gpr88 | Igfbp2 | Shisa6 | Slc15a3 |
| 5 | Pde1b | Efhd2 | Nrgn | Cldn11 | Csdc2 | Snap25 | Col1a2 | Nrgn | Ly6h | Mag | Pbx3 | Stmn1 | Adcy5 | Kl | Neurod2 | Map3k14 |
| 6 | Scn4b | Olfm1 | Olfm1 | Mag | Th | Cplx1 | Aldh1a2 | Lingo1 | Tmem130 | Plekhb1 | Ptpro | Lmo3 | Gng7 | Fxyd1 | Hpca | Tep1 |
| 7 | Adcy5 | Tbr1 | Enc1 | Cnp | Nmb | 3110035E14Rik | Atp1a2 | Olfm1 | Ndn | Mal | Cpne4 | Chgb | Pde10a | Lbp | Cck | AU021092 |
| 8 | Rasd2 | Stmn1 | Lingo1 | Plekhb1 | Gng4 | Adcy1 | Fmod | Nptxr | Gap43 | Pvalb | Ablim3 | Syp | Pcp4l1 | Trf | Cnih2 | Bmpr1b |
| 9 | Gng7 | Cck | Atp1a1 | Bcas1 | Shisa8 | Stmn1 | Ogn | Nrsn1 | Resp18 | Sparc | Pbx1 | Stmn2 | Inf2 | Folr1 | Prkca | Znhit6 |
Then, we annotated these spatial domains accroding to their marker genes and spatial locations (reference: Allen Brain Reference Atlases).
[13]:
cluster2annotation = {
'0': 'Striatum',
'1': 'Cortical layer6',
'2': 'Cortical layer2/3',
'3': 'Fiber tracts',
'4': 'Olfactory bulb(outer)',
'5': 'Cortical layer5',
'6': 'Cortical layer1',
'7': 'Cortical layer4',
'8': 'Hypothalamus',
'9': 'Thalamus',
'10': 'Olfactory bulb(inner)',
'11': 'Subplate',
'12': 'Striatum',
'13': 'Choroid plexus',
'14': 'Hippocampus',
'15': 'Others',
}
[14]:
adata.obs['annotation'] = adata.obs['leiden'].map(cluster2annotation).astype('category')
Spatial domain DE genes#
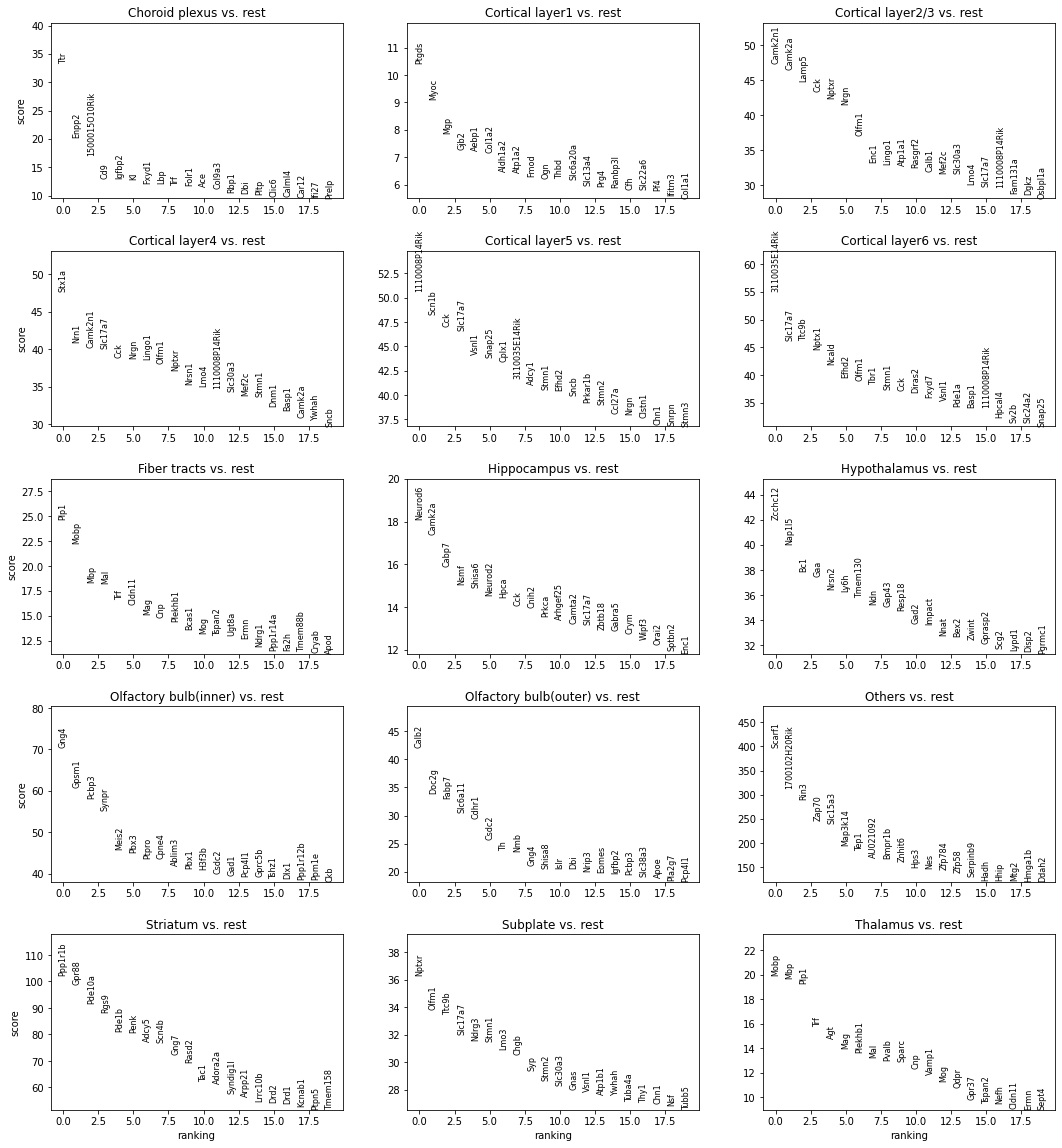
Lastly, we compute the differentially expressed (DE) genes of each annotated spatial domain for visualization.
[15]:
sc.tl.rank_genes_groups(adata, groupby='annotation', method='t-test')
[16]:
sc.pl.rank_genes_groups(adata, n_genes=20, ncols=3, sharey=False)
[17]:
DE_genes = pd.DataFrame(adata.uns['rank_genes_groups']['names']).iloc[:10,:]
DE_genes
[17]:
| Choroid plexus | Cortical layer1 | Cortical layer2/3 | Cortical layer4 | Cortical layer5 | Cortical layer6 | Fiber tracts | Hippocampus | Hypothalamus | Olfactory bulb(inner) | Olfactory bulb(outer) | Others | Striatum | Subplate | Thalamus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Ttr | Ptgds | Camk2n1 | Stx1a | 1110008P14Rik | 3110035E14Rik | Plp1 | Neurod6 | Zcchc12 | Gng4 | Calb2 | Scarf1 | Ppp1r1b | Nptxr | Mobp |
| 1 | Enpp2 | Myoc | Camk2a | Nrn1 | Scn1b | Slc17a7 | Mobp | Camk2a | Nap1l5 | Gpsm1 | Doc2g | 1700102H20Rik | Gpr88 | Olfm1 | Mbp |
| 2 | 1500015O10Rik | Mgp | Lamp5 | Camk2n1 | Cck | Ttc9b | Mbp | Cabp7 | Bc1 | Pcbp3 | Fabp7 | Rin3 | Pde10a | Ttc9b | Plp1 |
| 3 | Cd9 | Gjb2 | Cck | Slc17a7 | Slc17a7 | Nptx1 | Mal | Nsmf | Gaa | Synpr | Slc6a11 | Zap70 | Rgs9 | Slc17a7 | Trf |
| 4 | Igfbp2 | Aebp1 | Nptxr | Cck | Vsnl1 | Ncald | Trf | Shisa6 | Nrsn2 | Meis2 | Cdhr1 | Slc15a3 | Pde1b | Ndrg3 | Agt |
| 5 | Kl | Col1a2 | Nrgn | Nrgn | Snap25 | Efhd2 | Cldn11 | Neurod2 | Ly6h | Pbx3 | Csdc2 | Map3k14 | Penk | Stmn1 | Mag |
| 6 | Fxyd1 | Aldh1a2 | Olfm1 | Lingo1 | Cplx1 | Olfm1 | Mag | Hpca | Tmem130 | Ptpro | Th | Tep1 | Adcy5 | Lmo3 | Plekhb1 |
| 7 | Lbp | Atp1a2 | Enc1 | Olfm1 | 3110035E14Rik | Tbr1 | Cnp | Cck | Ndn | Cpne4 | Nmb | AU021092 | Scn4b | Chgb | Mal |
| 8 | Trf | Fmod | Lingo1 | Nptxr | Adcy1 | Stmn1 | Plekhb1 | Cnih2 | Gap43 | Ablim3 | Gng4 | Bmpr1b | Gng7 | Syp | Pvalb |
| 9 | Folr1 | Ogn | Atp1a1 | Nrsn1 | Stmn1 | Cck | Bcas1 | Prkca | Resp18 | Pbx1 | Shisa8 | Znhit6 | Rasd2 | Stmn2 | Sparc |
Now, we focus on a specific annotated spatial domain, here Striatum for demonstration.
We visualize the expression levels of the first-ranked DE gene of Striatum in UMAP space.
[18]:
fig, axs = plt.subplots(figsize=(8, 7))
sc.pl.umap(
adata,
color=DE_genes.iloc[0,12],
size=100,
cmap='summer',
vmin='p50',
vmax='p99',
show=False,
ax=axs,
)
plt.tight_layout()
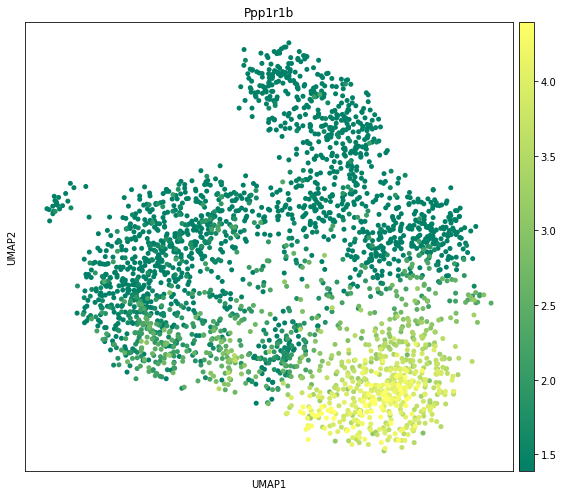
We also visualize the expression levels of the first-ranked DE gene of Striatum in spatial coordinates.
[19]:
fig, axs = plt.subplots(figsize=(8, 7))
sc.pl.spatial(
adata,
img_key='hires',
color=DE_genes.iloc[0,12],
size=1.5,
cmap='summer',
vmin='p50',
vmax='p99',
show=False,
ax=axs,
)
plt.tight_layout()
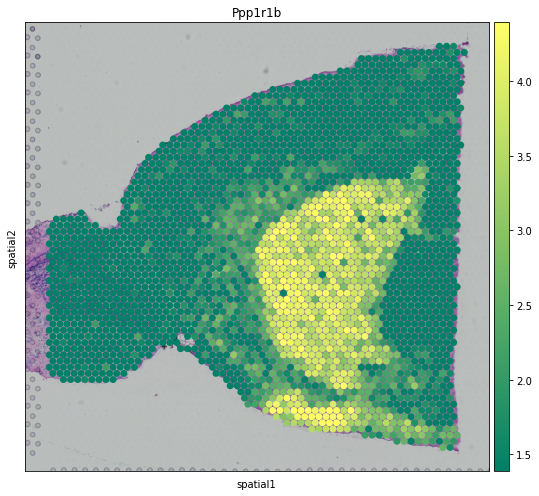
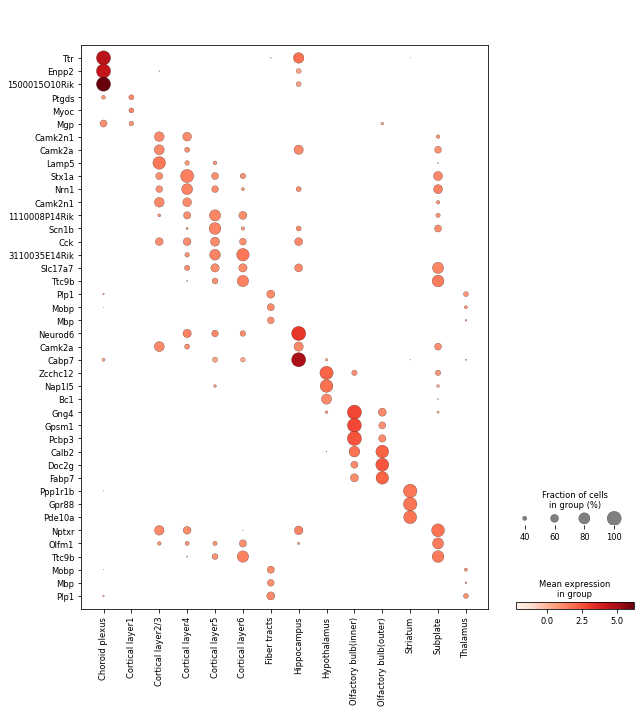
Then, we scale the data and visualize the expression pattern of top 3 DE genes of each annotated spatial domain.
[20]:
sc.pp.scale(adata)
[21]:
fig, axs = plt.subplots(figsize=(9, 10))
sc.pl.dotplot(
adata[adata.obs['annotation'] != 'Others', :],
var_names=DE_genes.drop('Others', axis=1).iloc[0:3, :].to_numpy().T.reshape(-1),
groupby='annotation',
expression_cutoff=1,
dot_min=0.25,
swap_axes=True,
show=False,
ax=axs,
)
plt.tight_layout()